Predicting Protein Folding with Deep Learning
-- posted December 2021 --
AphaFold is an AI method for predicting the 3-dimensional shape of proteins, and it is universally considered one of the most important recent scientific breakthroughs. AlphaFold was developed by DeepMind, the U.K.-based AI-focused subsidiary of Alphabet Inc., that is, Google.
As we know, proteins are essential building blocks of cells in all living organisms. Proteins represent sequences of amino acids; there are more than 20 different amino acids that can make up a protein. The 3-dimensional structures of proteins consist of many intricate folds, formed via complex interactions between the constituent amino acids. The figure shows the four principal structures of protein folding: primary structure (chain of amino acids), secondary structure (alpha helices and beta-pleated sheets), tertiary structure (3D repeated pattern), and quaternary structure (interactions of multiple amino acid chains).
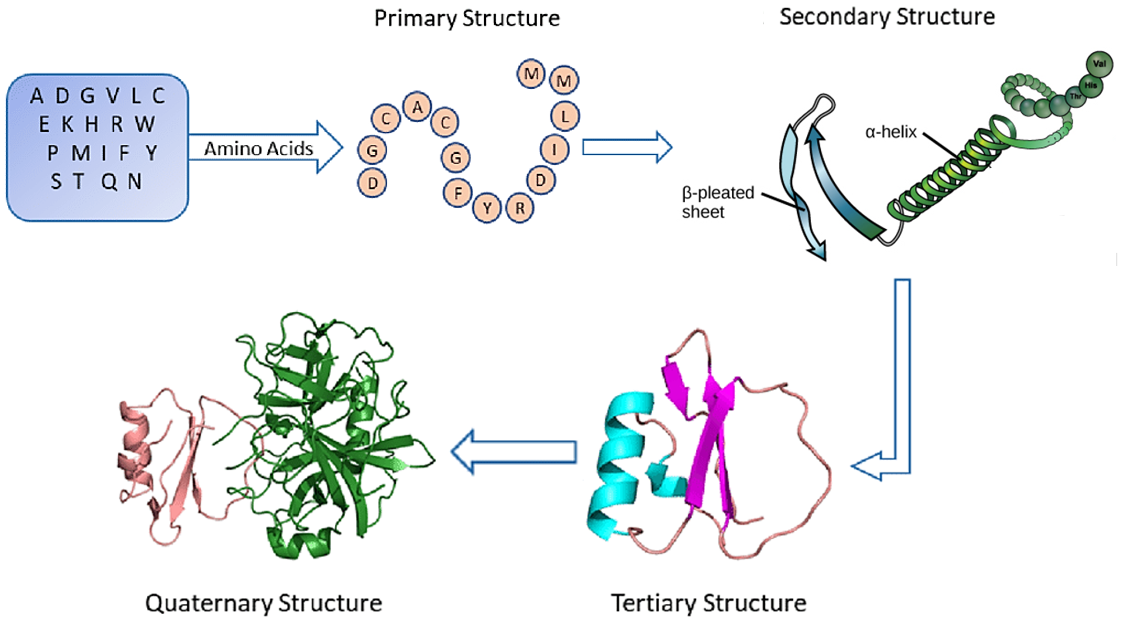
Figure: Four structures of protein folding. Source link.
Scientists in molecular biology have long observed that the 3D structure of proteins is directly related to their functionality. However, understanding how the sequential ordering and resulting interactions of amino acids impact the fold forming and the final spatial shape of a protein was for a long time beyond the capabilities of our predictive methods. This is referred to as the “protein folding problem,” and represents one of the grand challenges in biology.
Several laboratory methods are presently used to experimentally determine the 3D shape of proteins. X-ray crystallography entails recording the diffraction of X-rays when projected onto crystallized proteins. Another popular method that has been replacing X-ray crystallography is cryo-electron microscopy. This technique cools down samples to very low (cryogenic) temperatures and measures the scattering of a beam of applied electrons. Still, these laboratory methods are laborious, time-consuming, and expensive. As a result, although we know of over 200 million proteins across all living organisms, the 3D structure of only a small fraction of all proteins has been experimentally determined (about 400 thousand proteins).
To address the challenge of protein-structure predicting and motivate the development of new computational methods, a competition called CASP (Critical Assessment of Techniques for Protein Structure Prediction) has been organized biennially for over 25 years. CASP invites research teams to submit solutions for a target set of proteins (e.g., there were 97 target proteins in 2020), whereas a team of scientists compares the anonymized predictions for the 3D shapes of the proteins by the competing teams to the corresponding shapes obtained by laboratory experiments. A CASP score of 100 means that the predicted 3D shapes are identical matches to the experimentally obtained ones.
The figure below shows the best CASP scores since 2006. AlphaFold 1 by DeepMind was submitted to CASP in 2018 and outperformed existing methods by a substantial margin, achieving a median global distance CASP score of 58.9. An improved version AlphaFold 2 submitted to the 2020 challenge achieved even better performance, with a median CASP score of 87. Specifically, AlphaFold 2 obtained a score over 92 for more than half of the target proteins, as well as, 88% of the predicted 3D shapes had an average distance to the experimental shape of less than 4 Angstroms (where 1 Angstrom = 10-10 meters, and the radii of atoms range from 1A to 5A).
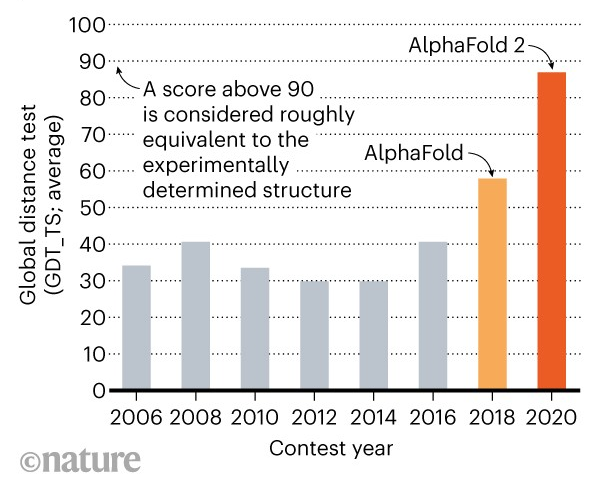
Figure: CASP global distance scores. Source link.
Both AlphaFold 1 and 2 employ deep learning neural networks for predicting the distance between the amino acids in proteins. Interestingly, the architecture of the neural layers in AlphaFold 2 is based on the attention-based transformer architectures, which have been initially developed for training large language models (e.g., GPT-3 is one of the most well-known such models). A high-level description of the block design of AlphaFold 2 is shown below.
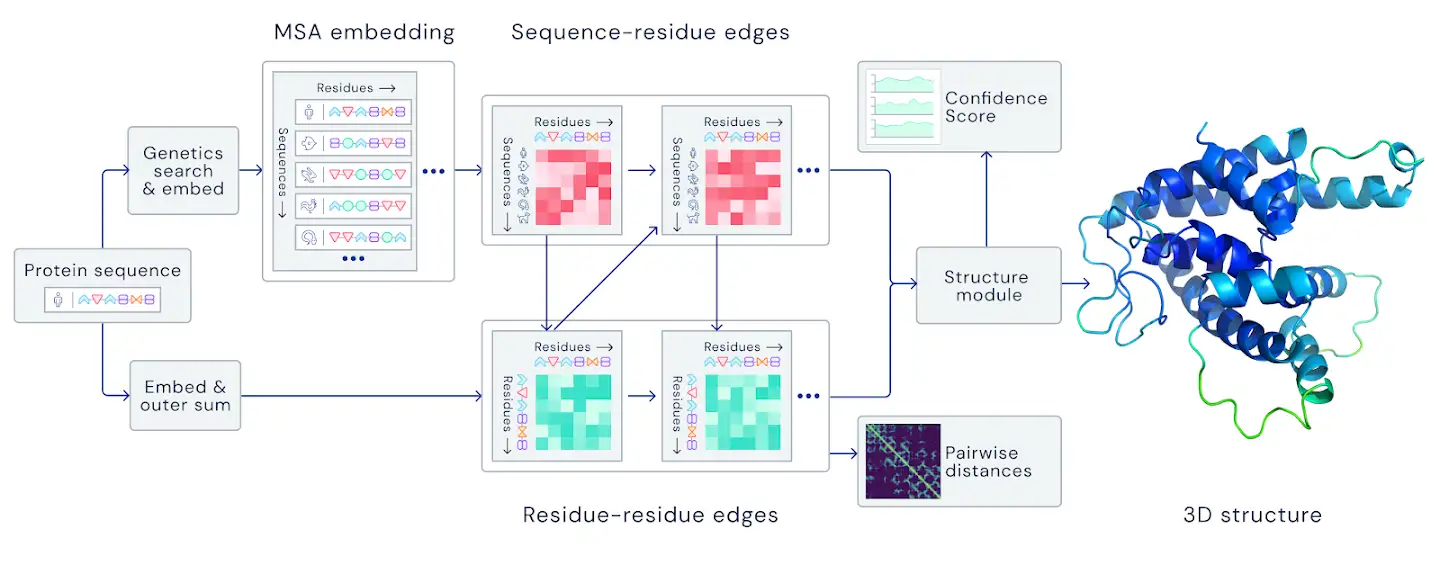
Figure: AlphaFold 2 block design is based on sequence-residue attention modules. Source link.
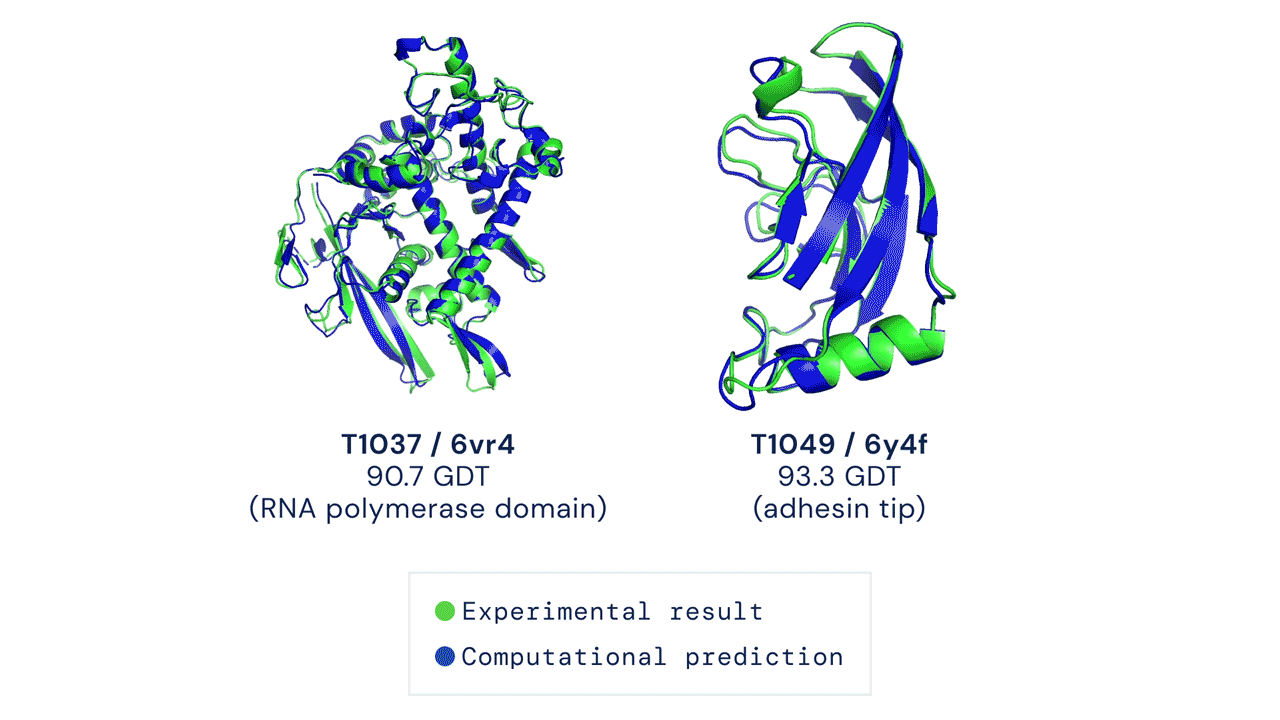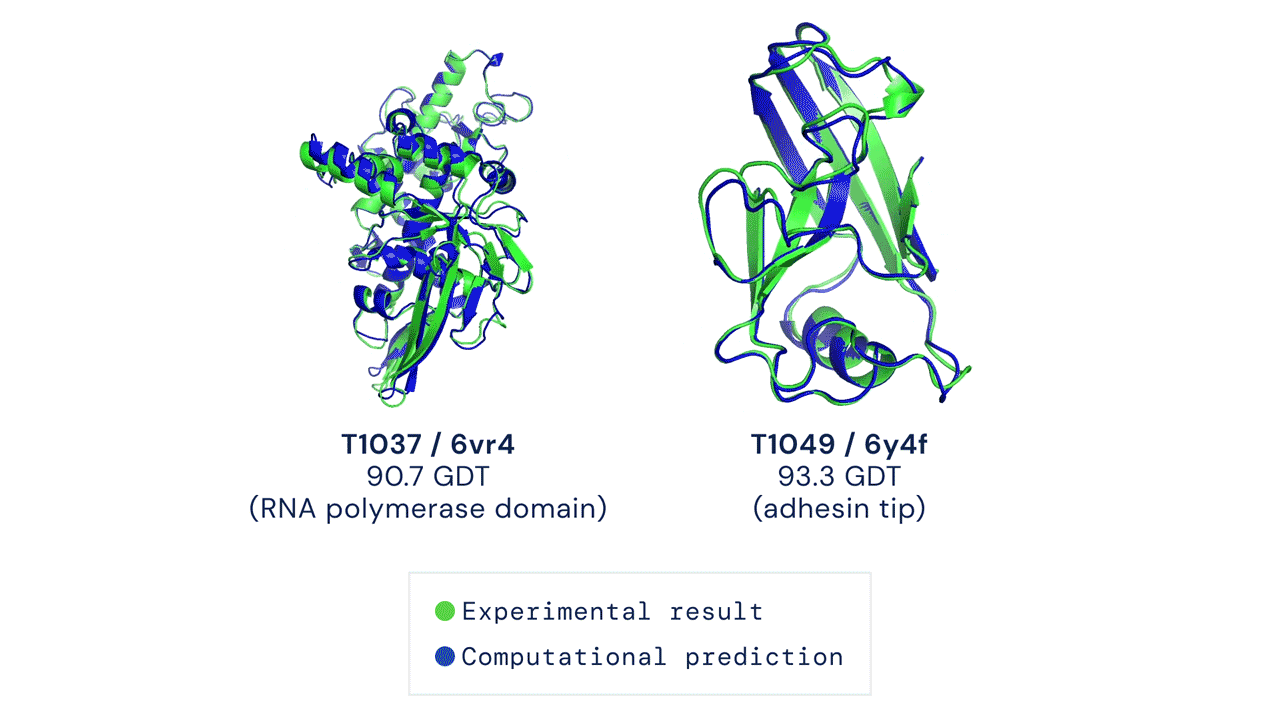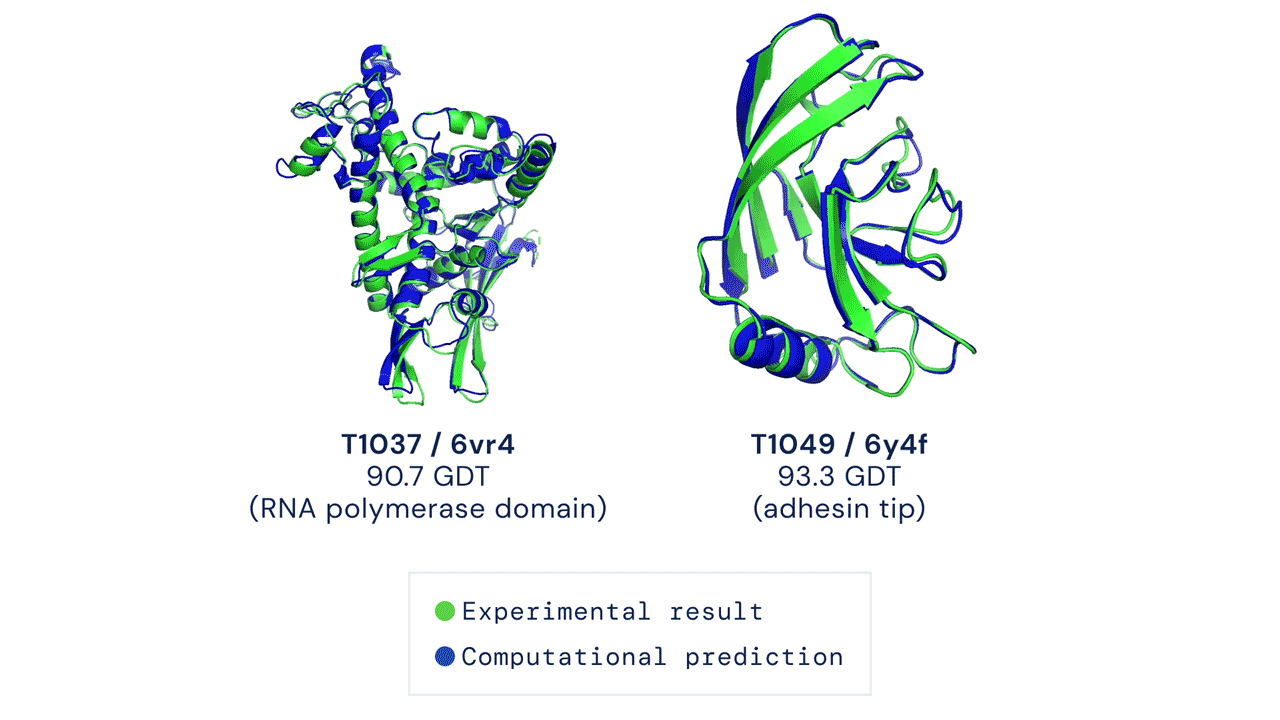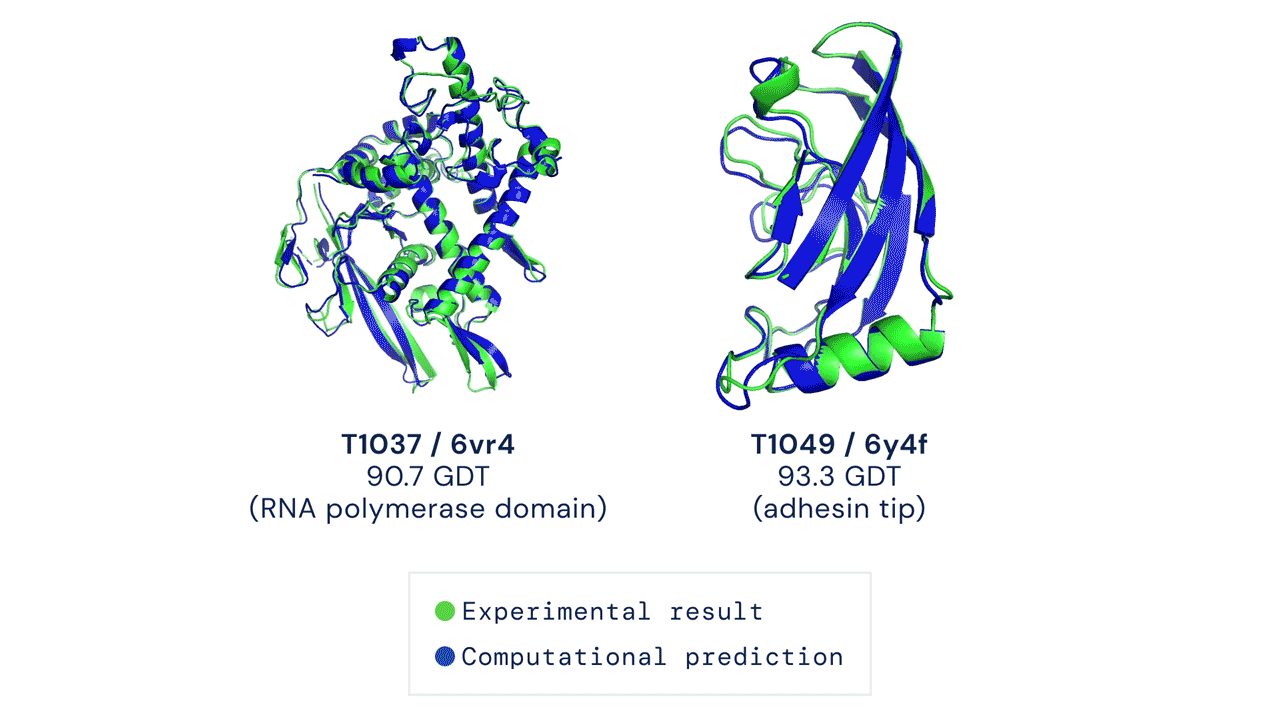
Figure: Experimentally obtained shape (green) and predicted shape by AlphaFold 2 (blue) for two proteins. Source link.
In 2021, DeepMind open-sourced the codes for AlphaFold 2 (link to the repository on GitHub). Also, they released a database consisting of over 360 thousand proteins (link), including the human proteome and proteomes of 20 organisms (proteome is the set of all expressed proteins in an organism).
Many scientists have described the accomplishment by AlphaFold as nothing short of transformational and sensational. The ability to accurately predict protein structures can revolutionize our understanding of protein functions in cell mechanisms, and in biology and medicine in general, and it can spur a wave of innovation in various medical applications, such as drug discovery and protein design.
Here are two videos that provide explanations of the protein folding prediction by AlphaFold: one short 1:51 minute video (link), and one longer 12:00 minute video (link).